jueves, 6 de junio de 2013
Preguntas más Frecuentes sobre Enfermedades Hereditarias
¿Existen pruebas de ADN que indican la predisposición a sufrir infartos?
Si. LabGenetics realiza una prueba de ADN, denominada "diagnóstico de trombofilia hereditaria", en la que se analizan diferentes mutaciones asociadas a la predisposición genética a padecer tromboembolismo venoso (trombosis), que ocurre de manera espontánea a edad temprana (antes de lso 40 años) sin causa aparente y con tendencia a recurrir.¿Puedo saber con un 100% de probabilidad si voy a desarrollar un cáncer?
LabGenetics realiza pruebas genéticas de ADN para analizar la presencia o ausencia de mutaciones en genes que confieren un incremento en el riesgo de desarrollar un cáncer. Por ejemplo, mutaciones en los genes BRCA1 y 2 aumentan el riesgo de una persona a desarrollar cáncer de mama y ovario. Sin embargo, es importante indicar que las personas con mutaciones en esos genes no necesariamente van a desarrollar cáncer.¿Existen pruebas genéticas de ADN para el diagnóstico precoz del Parkinson y el Alzheimer?
LabGenetics ofrece pruebas genéticas de ADN para la búsqueda de mutaciones en los genes relacionados con la aparición de las principales enfermedades neurológicas, incluyendo la enfermedad de Alzheimer y la de Parkinson.¿La infertilidad masculina puede tener causas genéticas?
Si. Está comprobado que determinadas alteraciones en el cromosoma Y están asociadas a la ausencia de espermatozoides en el semen (azoospermia) y a la disminución del número y movilidad de los espermatozoides (oligospermia). LabGenetics realiza pruebas genéticas de ADN para detectar microdelecciones en el cromosoma Y causantes de más del 20% de los casos de infertilidad no obstructiva, así como la detección de mutaciones en el gen CFTR causantes de la denominada "ausencia bilateral congénita de conductos deferentes".¿Es posible realizar algún tipo de prueba genética de ADN a mujeres con abortos espontáneos recurrentes?
Si. En el caso de mujeres con problemas de abortos espontáneos recurrentes de causa desconocida, o con problemas de preeclampsia o eclampsia, es recomendable realizar un estudio genético para descartar la presencia de mutaciones en genes relacionados con el mecanismo de coagulación (Factor V de Leiden, Factor II y MTHFR). Consúltenos¿Es posible conocer cómo va a responder una persona ante un determinado medicamento?
Si, en algunos casos. LabGenetics es uno de los pocos laboratorios españoles que posee una línea de servicios dedicada a la farmacogenética, en la que se analizan diferentes polimorfismos (variabilidad genética) en genes asociados al metabolismo de diferentes medicamentos, con el objetivo de poder ofrecer un tratamiento terapéutico lo más personalizado posible. Consulte aquí las aplicaciones de nuestro servicio de farmacogenéticaPeliculas sobre enfermedades Geneticas
El aceite de lorenzo : http://www.peliculasmas.com/tag/aceite-de-lorenzo/
El hombre elefante: http://pelis24.com/pelicula-ca/14275-el-hombre-elefante-1980.html
Medidas desesperadas: http://pelis24.com/pelicula-ca/14275-el-hombre-elefante-1980.html
El hombre elefante: http://pelis24.com/pelicula-ca/14275-el-hombre-elefante-1980.html
Medidas desesperadas: http://pelis24.com/pelicula-ca/14275-el-hombre-elefante-1980.html
miércoles, 5 de junio de 2013
Hemofilia
Definición
Es una enfermedad genética ligada al cromosoma X que consiste en la dificultad de la sangre para coagularse adecuadamente. Se caracteriza por la aparición de hemorragias internas y externas debido a la deficiencia parcial de una proteína coagulante denominada globulina antihemofílica (factor de coagulación).
 Los
factores de coagulación (enumeradas I, II,..., XIII) son un grupo de
proteínas, que al producirse una hemorragia, son las responsables de
activar el proceso de coagulación. Se caracterizan por actuar en forma
de cascada, es decir, uno activa al siguiente.
Los
factores de coagulación (enumeradas I, II,..., XIII) son un grupo de
proteínas, que al producirse una hemorragia, son las responsables de
activar el proceso de coagulación. Se caracterizan por actuar en forma
de cascada, es decir, uno activa al siguiente.
Cuando
hay carencia de algún factor de coagulación, la sangre tarda más tiempo
en formar el coágulo y, aunque llegue a formarse, no es consistente y
no se forma un buen tapón para detener la hemorragia.
Tipos de hemofilia
Hay
dos tipos de hemofilia que se caracterizan ambas por la falta parcial o
total de una de las proteínas de la sangre que controlan las
hemorragias. Así tenemos:
-Hemofilia A: debido a una deficiencia del factor VIII. Afecta aproximadamente a 1 en 6.000 personas.
-Hemofilia B: debido a una deficiencia del factor IX. Afecta aproximadamente a 1 en 30.000 personas.
La gravedad de ambas hemofilias viene determinada por el nivel de actividad de coagulación del factor VIII o del factor IX en la sangre. De esta manera podemos dividir la hemofilia en:
-Hemofilia A: debido a una deficiencia del factor VIII. Afecta aproximadamente a 1 en 6.000 personas.
-Hemofilia B: debido a una deficiencia del factor IX. Afecta aproximadamente a 1 en 30.000 personas.
La gravedad de ambas hemofilias viene determinada por el nivel de actividad de coagulación del factor VIII o del factor IX en la sangre. De esta manera podemos dividir la hemofilia en:
* Leve:
presentan entre un 5% - 40% de factor si se compara con una persona
sana. Por lo general sólo sufren de hemorragias a consecuencia de
cirugías o lesiones graves, podrían nunca llegar a tener un problema
hemorrágico.
* Moderado:
presenta entre el 1% - 5% del normal. Estos padecen hemorragias con
menos frecuencia, generalmente después de una lesión y suelen
experimentar hemorragias espontáneas.
* Grave:
presentan menos del 1% del nivel normal. Padecen de hemorragias
espontáneas frecuentes en músculos o articulaciones. Sin tratamiento
preventivo, pueden sangrar una o dos veces por semana.
.
Diagnóstico
El
diagnóstico de la hemofilia se hace mediante la historia clínica y un
análisis de sangre para la medición, a través de pruebas especiales de
coagulación, de los grados de los diferentes factores. El objetivo es
establecer la severidad de la enfermedad y decidir el tratamiento más
adecuado a seguir.
Síntomas
El
síntoma más común de la hemofilia es la hemorragia incontrolable y
excesiva por causa del factor de coagulación que falta o está en bajos
niveles. Puede producirse una hemorragia incluso cuando no haya ninguna
lesión. Entre los síntomas más frecuentes están:

* Aparición de hematomas extensos por pequeños accidentes.
* Tendencia a sangrar fácilmente por la nariz, la boca y las encías por un traumatismo sin importancia.
*
Hemorragia en los músculos, el cual puede causar hinchazón, dolor y
enrojecimiento. La hinchazón por el exceso de sangre en estas zonas
puede producir un aumento de la presión en los tejidos y nervios de la
zona, provocando daño y, o deformación permanente.

*
Padecimiento de hemartrosis, que es la manifestación clínica mas
frecuente en los hemofílicos, consiste en el sangrado intraarticular que
afecta especialmente a las articulaciones de un solo eje como
la rodilla, el codo o el tobillo. Si se produce una hemartrosis en
repetidas ocasiones en una articulación, se origina una deformidad y
atrofiación muscular llamada artropatía hemofílica

* Hemorragia cerebral, el cual es la causa más común de muerte en personas hemofílicas. Entre los signos de este tipo de
hemorragia incluye cambios en el comportamiento, excesiva somnolencia,
dolores de cabeza, visión doble, vómitos, y convulsiones.
* Otras fuentes de hemorragia, como puede ser en la garganta, riñones, heces, orina (hematuria), ojo, etc.
Causas
Es
una enfermedad genética que se transmite de padres a hijos. El análisis
de los árboles genealógicos en familias hemofílicas, demostró que esta
enfermedad sólo se manifestaba en los varones relacionados por vía
materna y que, por tanto, debería consistir en una anomalía hereditaria
que se producía por algún defecto en el cromosoma X (herencia ligada al
sexo).
Las
instrucciones necesarias para ordenar a las células cómo fabricar las
proteínas que el organismo requiere para su funcionamiento, se
encuentran contenidas en pequeñas formaciones llamadas genes, dentro de
los cromosomas.
Los cromosomas vienen en pares, por lo que tenemos dos copias de todos nuestros genes, es decir, si hay algún daño en algún gen, hay una copia de respaldo. Pero hay una excepción, los cromosomas sexuales: X y Y. El cromosoma X contiene muchos genes que son comunes a ambos sexos, como los genes para la producción del factor VIII y el factor IX. La mujer tiene dos copias de esos genes específicos mientras que los varones sólo uno. Si el varón hereda un cromosoma con un gen dañado del factor VIII o IX, es el único gen que recibe y no tiene información de respaldo para producir ese factor de coagulación.

La
mujer se comportará como portadora de la enfermedad y el hombre la
padece (y transmite a la descendencia). La mujer para manifestar la
enfermedad necesitaría dos copias defectuosas, cosa muy poco probable.
En un 20% de los casos de hemofilia, el trastorno es causado por una mutación espontánea del gen.
Tratamiento
La
hemofilia se puede tratar mediante la administración por vía
intravenosa del factor deficiente VIII o IX a la dosis adecuada en
función de la edad y grado de severidad del episodio hemorrágico. Los
concentrados utilizados de factores pueden ser plasmáticos o
recombinantes (que no están hechos con sangre humana), ambos sometidos a
procesos de inactivación viral.
En algunos
casos los pacientes producen una respuesta inmune contra el factor
exógeno administrado. A estos pacientes que desarrollan de esta manera
los llamados “inhibidores”, se tratan con concentrados de mezclas de
factores de la coagulación.
También es
recomendada al paciente la práctica de deportes que ayudan al
fortalecimiento de los músculos y por lo tanto disminuir el riesgo de
hemorragias espontáneas, evitar la toma de aspirinas o ácido
acetilsalicílico, no someterse a inyecciones intramusculares, etc.
La
ventaja hoy día del tratamiento es que se puede llevar a cabo en el
propio domicilio mediante protocolos de autotratamiento por parte del
propio paciente.
Fenilcetonuria
La fenilcetonuria o PKU (del inglés
“phenylketonuria”) es un trastorno hereditario que afecta la química del
organismo y que provoca retraso mental. Afortunadamente, gracias a las
pruebas de detección precoz, es posible diagnosticar y tratar desde el
comienzo a casi todos los bebés recién nacidos afectados por este
trastorno y permitir que crezcan y se desarrollen con inteligencia
normal.
En los EE.UU., al menos uno de cada 25,000 bebés nace con PKU. Este trastorno se produce en todos los grupos étnicos, si bien es más común en las personas cuyos antepasados provienen del norte de Europa o fueron indígenas nativos de los EE.UU. que en personas de origen afroamericano, hispano y asiático.

En los EE.UU., al menos uno de cada 25,000 bebés nace con PKU. Este trastorno se produce en todos los grupos étnicos, si bien es más común en las personas cuyos antepasados provienen del norte de Europa o fueron indígenas nativos de los EE.UU. que en personas de origen afroamericano, hispano y asiático.
.¿Qué es la PKU?

Las
personas con PKU no pueden procesar una parte de la proteína
fenilalanina, que está presente en la mayoría de los alimentos. Debido a
una anomalía genética, las personas afectadas carecen de una enzima
(fenilalanina hidroxilasa o PAH) que convierte la fenilalanina en otras
sustancias que necesita el organismo, o bien la tienen pero en niveles
demasiado bajos. Si no se proporciona el tratamiento adecuado, la
fenilalanina va acumulándose en el torrente sanguíneo y produce daños
cerebrales y retraso mental.
2. ¿Cómo afecta la PKU a los niños?
2. ¿Cómo afecta la PKU a los niños?
Durante
sus primeros meses de vida, los niños nacidos con PKU parecen normales,
pero al llegar a la edad de un año, es evidente que padecen un retraso
en el desarrollo. Los niños con PKU sin tratar suelen ser irritables y
tener problemas de conducta. Pueden producir un olor similar al del moho
o la humedad y tener la piel seca, erupciones o convulsiones.
Habitualmente presentan un buen desarrollo físico y tienden a tener el
cabello más claro que sus hermanos.
¿Quiénes sufren de PKU?
Cuando
ambos padres son portadores, existe una probabilidad del 25% de que
ambos transfieran un gen PAH anormal a su bebé, haciendo que este nazca
con PKU, una probabilidad del 50% de que el bebé herede un gen PAH
anormal y el gen normal del otro, lo que lo convertiría en portador al
igual que sus padres, y una probabilidad de una en cuatro de que ambos
padres transmitan al bebé el gen normal y que éste no tenga la
enfermedad ni sea portador. Estas probabilidades son iguales en todos
los embarazos.
Un ''portador'' tiene un gen PAH normal y un gen PAH que contiene una mutación. La salud de los portadores no sufre efecto alguno por la presencia de este gen.
Un ''portador'' tiene un gen PAH normal y un gen PAH que contiene una mutación. La salud de los portadores no sufre efecto alguno por la presencia de este gen.

¿En qué consiste la prueba de detección PKU?
Se
practica una pequeña punción en el talón del bebé para extraer unas
pocas gotas de sangre. Por lo general, se envía la muestra a un
laboratorio médico regional para determinar si la cantidad de
fenilalanina es mayor a la normal y los resultados se envían al médico a
cargo del cuidado del bebé. Si los resultados no son normales, se
procede a la realización de pruebas adicionales para determinar si el
bebé tiene PKU o si su alto nivel de fenilalanina se debe a alguna otra
causa.

En ocasiones, es posible que un caso de PKU pase inadvertido si el análisis se realiza antes de las 24 horas de vida del bebé. En estos casos algunos expertos recomiendan repetir el análisis al bebé después de una o dos semanas de vida.
¿Es posible prevenir los síntomas de la PKU?
Sí.
Es posible prevenir el retraso mental si comienza a tratarse al bebé
con una dieta especial baja en fenilalanina lo antes posible después de
su nacimiento.

Al comienzo, se alimenta al bebé utilizando una fórmula especial que contiene proteínas, pero sin fenilalanina. Luego se añaden a su dieta ciertas verduras, frutas, algunos granos y otros alimentos con poca fenilalanina, pero nunca puede alimentárselo con leche normal, queso, huevos, carne, pescado ni otros alimentos de alto contenido proteico. Como las proteína
 s
son esenciales para el desarrollo y el crecimiento normales del niño,
éste debe continuar ingiriendo una de las fórmulas especiales que
contenga muchas proteínas y los nutrientes básicos pero que tenga muy
poca fenilalanina. Las bebidas y alimentos dietéticos que contienen el
edulcorante artificial aspartamo (Equal) están estrictamente prohibidos.
s
son esenciales para el desarrollo y el crecimiento normales del niño,
éste debe continuar ingiriendo una de las fórmulas especiales que
contenga muchas proteínas y los nutrientes básicos pero que tenga muy
poca fenilalanina. Las bebidas y alimentos dietéticos que contienen el
edulcorante artificial aspartamo (Equal) están estrictamente prohibidos.Es necesario realizar un seguimiento de los niños y los adultos con PKU en un centro médico que se especialice en este trastorno. La dieta debe ser individual, según la cantidad de fenilalanina que cada uno pueda tolerar. Todas las personas afectadas con este trastorno deben realizarse análisis de sangre periódicamente para medir los niveles de fenilalanina.
¿Qué es la PKU materna?
Se
estima que existen unas 3,000 mujeres en edad fecunda que han sido
tratadas exitosamente de PKU en los EE.UU. La mayoría de ellas abandonó
su dieta especial durante la niñez, porque en esa época la mayoría de
los médicos creía que hacerlo no conllevaba riesgos.

Cuando estas jóvenes quedan embarazadas mientras comen una dieta normal, la concentración de fenilalanina en su sangre es muy elevada. Los niveles elevados de fenilalanina en la sangre materna durante el embarazo pueden causar problemas serios al feto. En aproximadamente el 90% de estos casos, los bebés sufren retraso mental y cerca del 70% nace con una cabeza de tamaño reducido (microcefalia). Muchos de ellos también nacen con defectos cardíacos y bajo peso. Como la mayoría de estos bebés no hereda la PKU sino que su daño cerebral se debe íntegramente a los elevados niveles de fenilalanina de la madre durante el embarazo, la dieta para PKU no les resulta beneficiosa.
Afortunadamente, hay una manera de ayudar a prevenir el retraso mental y otros problemas que ocurren en los bebés de mujeres con PKU. Estas mujeres deben reanudar su dieta especial al menos tres meses antes del embarazo y continuarla durante el mismo. El Estudio Internacional sobre la PKU Materna descubrió que las mujeres cuyos niveles de fenilalanina en la sangre se encontraban bajo control antes de la concepción, o después de 8 a 10 semanas de embarazo a lo sumo, tenían las mismas probabilidades de tener bebés sanos que las mujeres sin PKU.
¿Se pueden usar medicamentos para manejar la PKU?
En diciembre del 2007, la Food and Drug Administration (FDA) aprobó Kuvan (sapropterin dihydrocholoride), el primer medicamento para ayudar a manejar la PKU. El medicamento ayuda a reducir los niveles de fenilalanina en la sangre en individuos con PKU al aumentar la actividad de la encima PAH.Kuvan es efectivo solamente en individuos que tienen cierta actividad de la PAH. Las personas que toman este medicamento deben continuar siguiendo una dieta especial baja en fenilalanina y recibir análisis de sangre para medir los niveles de fenilalanina.
¿Qué novedades hay en las investigaciones sobre la PKU?
Los investigadores están estudiando los beneficios de un suplemento nutricional llamado BH4 en las personas con PKU. Otros están desarrollando una versión genéticamente manipulada de la enzima faltante. Ambos enfoques podrían permitir a las personas afectadas alimentarse con una dieta casi normal. Los investigadores también están analizando la posibilidad de tratar la PKU mediante terapia genética.Síndrome de Klinefelter
Definición
Es una anomalía cromosómica que afecta solamente a los hombres y ocasiona hipogonadismo.
El sexo de las personas está determinado por los cromosomas X e Y. Los hombres tienen los cromosomas XY y las mujeres tienen los cromosomas XX. En el síndrome de Klinefelter se presentan los cromosomas XXY.
Esta condición es común y afecta a 1 en 500 hombres. Al nacer, el niño presenta una apariencia normal, pero el defecto usualmente comienza a notarse cuando llega a la pubertad y las características sexuales secundarias no se desarrollan o lo hacen de manera tardía.
Genética
El síndrome de Klinefelter se considera la anomalía gonosómica más común en los humanos, presentándose con una incidencia de 1 en 500 en los recién nacidos vivos varones. Los afectados presentan un cromosoma “X” supernumerario lo que conduce a fallo testicular primario con infertilidad e hipoandrogenismo. A pesar de la relativa frecuencia del padecimiento en recién nacidos vivos, se estima que la mitad de los productos 47, XXY se abortan de manera espontánea.

Fue descrito en 1942 por Klinefelter y colaboradores, estudiaron 9 pacientes con: ginecomastia, testículos pequeños, azoospermia, cifras elevadas de gonadotropinas. Ellos sugirieron que el defecto primario estaba en las células de Sertoli y propusieron que además en estos pacientes había una deficiencia en una hormona testicular que regulaba la concentración de gonadotropinas hipofisiarias, a la que llamaron hormona “X” o inhibina.

En 1956 se demostró la presencia del corpúsculo de Barr en pacientes con síndrome de Klinefelter y en 1959 se identifica que el cariotipo de un sujeto con la enfermedad era 47, XXY. De esta manera se estableció que la presencia de un cromosoma “X” extra era el factor etiológico fundamental.

Síntomas
* Tejido mamario agrandado (ginecomastia).
* Testículos poco desarrollados.
* Azoospermia
* Cifras elevadas de gonadotropinas
* Pene pequeño.
* Vello púbico, axilar o facial disminuido.
* Disfunción sexual.
* Estatura alta (pubertad).
* Discapacidad para el aprendizaje.
* Eyaculación precoz
* Testículos poco desarrollados.
* Azoospermia
* Cifras elevadas de gonadotropinas
* Pene pequeño.
* Vello púbico, axilar o facial disminuido.
* Disfunción sexual.
* Estatura alta (pubertad).
* Discapacidad para el aprendizaje.
* Eyaculación precoz
Causas
Nadie sabe qué es lo que pone a una pareja en riesgo de concebir un niño XXY. La edad avanzada de la madre aumenta el riesgo de cromosoma XXY, pero solo un poco. Estudios recientes enseña que la mitad de las veces, el cromosoma extra proviene del padre.Se explica que las células destinadas para la producción de esperma u óvulos pasan por un proceso conocido como meiosis. En este proceso, los 46 cromosomas en la célula se separan, produciendo dos células nuevas que tienen 23 cromosomas cada una. Pero, antes que la meiosis se cumpla, los cromosomas se emparejan con sus cromosomas correspondientes e intercambian piezas de material genético.

En algunos casos, los cromosomas fallan en emparejarse e intercambiar material genético. En ocasiones, esto resulta en el movimiento de ambos a la misma célula produciendo un huevo con dos “X” o el esperma que tiene cromosomas “X” e “Y”. Cuando el esperma tiene ambos cromosomas fertiliza un huevo que tiene un cromosoma singular de “X” o un esperma normal que tiene “Y” fertiliza un huevo que tiene dos cromosomas “X”, un varón “XXY” es concebido.
TratamientoEl tratamiento consiste en una terapia con testosterona, el cual puede lograr lo siguiente:
- Mejorar la fuerza

- Mejorar la apariencia de los músculos
- Promover el crecimiento de vello corporal
- Mejorar la autoestima y el estado de ánimo
- Mejorar la energía y el impulso sexual
- Mejorar la concentración
La mayoría de los pacientes no pueden engendrar hijos. Sin embargo, hay algunos casos de hombres con un cromosoma X extra que tienen descendencia sana, algunas veces con la ayuda de especialistas en infertilidad.
Síndrome de Cri-du-chat
Definición
El síndrome del maullido de gato (del francés Cri-du-Chat), es una enfermedad congénita infrecuente con alteración cromosómica provocada por un tipo de deleción estructural del brazo corto del cromosoma 5, caracterizada por un llanto que se asemeja al maullido de un gato.
El síndrome del maullido fue descrito inicialmente por Lejeune en 1963. Tiene una prevalencia estimada de aproximadamente de 1 en 20.000 nacimientos y predomina en las niñas

Causas
La
causa del síndrome del maullido del gato es la supresión de genes en el
cromosoma 5. Uno de los genes suprimidos llamado telomerasa
transcriptasa inversa (TERT) y dependiendo del tamaño de la porción
perdida la afección será mayor o menor. La causa de esta rara supresión
cromosómica se desconoce, pero se cree que entre el 85%-90% de los casos
se debe a la pérdida espontánea de una parte del cromosoma 5 durante el
desarrollo de un óvulo o de un espermatozoide. En un 10%-15% de estos
casos se debe a que uno de los padres es portador de una reorganización
del cromosoma 5 denominada translocación.
Síntomas 

* La cara suele ser redondeada, llena y mofletuda, con el paladar elevado y escarpado.
* Cabeza pequeña (microcefalia).
* Inclinación de los ojos hacia abajo
* Ojos separados (hipertelorismo).
* Orejas de implantación baja (pueden estar malformadas).
* Quijada pequeña (micrognatia).
* Mala oclusión dental.
* Llanto de tono alto similar al de un gato, debido al anormal desarrollo de la glotis y laringe.
* Bajo peso al nacer y crecimiento lento.
* Un solo pliegue en la palma de la mano (pliegue simiesco)
* Desarrollo lento o incompleto de las habilidades motoras.
* Retraso mental.
* Miopía y atrofia óptica
* Pies planos.
* Metacarpianos y metatarsianos pequeños.
* Pliegue del epicanto (pliegue extra de piel sobre el ángulo interior del ojo)
* Cabeza pequeña (microcefalia).
* Inclinación de los ojos hacia abajo
* Ojos separados (hipertelorismo).
* Orejas de implantación baja (pueden estar malformadas).
* Quijada pequeña (micrognatia).
* Mala oclusión dental.
* Llanto de tono alto similar al de un gato, debido al anormal desarrollo de la glotis y laringe.

* Bajo peso al nacer y crecimiento lento.
* Un solo pliegue en la palma de la mano (pliegue simiesco)
* Desarrollo lento o incompleto de las habilidades motoras.
* Retraso mental.
* Miopía y atrofia óptica
* Pies planos.
* Metacarpianos y metatarsianos pequeños.
* Pliegue del epicanto (pliegue extra de piel sobre el ángulo interior del ojo)
Tratamiento
 No
hay un tratamiento específico disponible para este síndrome. Se debe
abordar el retardo mental y se recomienda la asesoría para los padres.
No
hay un tratamiento específico disponible para este síndrome. Se debe
abordar el retardo mental y se recomienda la asesoría para los padres.Los padres de un niño con este síndrome deben buscar asesoría genética y someterse a una prueba de cariotipo con el fin de determinar si uno de ellos tiene una reordenación del cromosoma 5
Pronóstico
El
pronóstico varía, pero lo usual es el retardo mental. La mitad de los
niños afectados aprende habilidades verbales suficientes para
comunicarse, el llanto similar a un gato se vuelve menos evidente a
medida que pasa el tiempo, los cambios en la pubertad serán típicos,
etc.
Pueden presentar complicaciones como incapacidad de valerse por sí solo e incapacidad de desenvolverse socialmente.
Pueden presentar complicaciones como incapacidad de valerse por sí solo e incapacidad de desenvolverse socialmente.
Síndrome de Turner
Definición
El
síndrome de Turner es una enfermedad genética caracterizada por
presencia de un solo cromosoma X. Fenotípicamente son mujeres (por
ausencia de cromosoma Y). Este trastorno inhibe el desarrollo sexual y
causa infertilidad. Su incidencia es de alrededor de 1 en cada 2.500
niñas.
Usualmente es esporádico, lo que significa que no es heredado de uno de los padres. En pocos casos, uno de los padres lleva cromosomas reorganizados que pueden ocasionar el síndrome de Turner en una hija; esta es la única situación en la que este síndrome es heredado. La condición se diagnóstica ya sea al nacer o en la pubertad cuando existe ausencia o retraso de la menstruación y se presenta un retraso en el desarrollo de las características sexuales secundarias normales.
El nombre "síndrome de Turner" proviene del médico Dr. Henry Turner, quien fue el primero en describir el conjunto de descubrimientos en 1938. No fue sino hasta 1959 que se identificó la causa del síndrome de Turner (la presencia de un sólo cromosoma X).
Usualmente es esporádico, lo que significa que no es heredado de uno de los padres. En pocos casos, uno de los padres lleva cromosomas reorganizados que pueden ocasionar el síndrome de Turner en una hija; esta es la única situación en la que este síndrome es heredado. La condición se diagnóstica ya sea al nacer o en la pubertad cuando existe ausencia o retraso de la menstruación y se presenta un retraso en el desarrollo de las características sexuales secundarias normales.
El nombre "síndrome de Turner" proviene del médico Dr. Henry Turner, quien fue el primero en describir el conjunto de descubrimientos en 1938. No fue sino hasta 1959 que se identificó la causa del síndrome de Turner (la presencia de un sólo cromosoma X).
Síntomas 

* Talla baja
* Falla gonadal (infertilidad)
* Falla gonadal (infertilidad)
* Micrognatia (falta de desarrollo mandibular)
* Implantación baja del pelo
* Paladar ojival
* Cuarto metacarpiano corto
* Escoliosis
* Rasgos oculares anormales (caída de los párpados)
* Desarrollo óseo anormal
* Implantación baja del pelo
* Paladar ojival
* Cuarto metacarpiano corto
* Escoliosis
* Rasgos oculares anormales (caída de los párpados)
* Desarrollo óseo anormal
* Mamas pequeñas y vello púbico disperso
* Lagrimeo disminuido
* Menstruación ausente
* Pliegue simiesco (un sólo pliegue en la palma)
* Carencia de la humedad normal en la vagina, relaciones sexuales dolorosas
* Lagrimeo disminuido
* Menstruación ausente
* Pliegue simiesco (un sólo pliegue en la palma)
* Carencia de la humedad normal en la vagina, relaciones sexuales dolorosas
Signos y exámenes

- Cariotipo X0, presente en el síndrome de Turner. El examen físico revela genitales y mamas subdesarrollados, cuello corto, baja estatura y desarrollo anormal del tórax.
- El cariotipo muestra 45 cromosomas con un modelo de 44 X, o es decir, un cromosoma sexual ausente.
- El ultrasonido puede revelar órganos reproductores femeninos pequeños o subdesarrollados.
- El examen ginecológico puede revelar sequedad del recubrimiento de la vagina.
- La hormona luteinizante y foliculoestimulante sérica se encuentra elevada.
Complicaciones
* Presión sanguínea alta (hipertensión)
* Obesidad
* Diabetes mellitus
* Tiroiditis de Hashimoto
* Cataratas
* Artritis
Tratamiento
La hormona del crecimiento puede ayudar a una niña con síndrome de Turner a incrementar su estatura. La terapia con reemplazo de estrógenos con frecuencia se inicia cuando la niña tiene 12 ó 13 años de edad y ayuda a estimular el crecimiento de las mamas, del vello púbico y otras características sexuales.
Las mujeres con este síndrome que deseen quedar en embarazo pueden pensar en la utilización de un óvulo de donante.
Síndrome de Edwards
Definición
El Sindrome de Edwards, más conocida como trisomía 18. Es una aneuploidía humana que se caracteriza por la presencia de un cromosoma adicional en el par 18. Fue originalmente descrita por John H. Edwards en la Universidad de Wisconsin, sus resultados fueron publicados y registrados en la literatura pediátrica y genética en el año 1960. Los
estudios de genética molecular, no han descrito con claridad las
regiones puntuales que necesitan ser duplicadas para que se produzca el
fenotipo característico del síndrome Edwards. Hasta el momento solo se
conocen dos regiones del brazo largo: 18q12-21 y 18q23.
Los
estudios de genética molecular, no han descrito con claridad las
regiones puntuales que necesitan ser duplicadas para que se produzca el
fenotipo característico del síndrome Edwards. Hasta el momento solo se
conocen dos regiones del brazo largo: 18q12-21 y 18q23.Debido a su alta tasa de mortalidad en los recién nacidos, (90% de los casos) se le ha considerado como una enfermedad de tipo “letal”
2. Epidemiología
La
enfermedad se ha descrito con mayor frecuencia en embarazos de mujeres
de edad avanzada por sobre los 35 años. No existe evidencia respecto a
que se su prevalencia este relacionada con razas o zonas geográficas en
particular.
Investigaciones posteriores sugieren que el origen de la trisomia 18 es la no disyunción de los cromosomas durante la meiosis o mitosis postcigotica. Durante la investigación se observo que alrededor del 50% de los errores en la separación de los cromosomas en la ovogénesis, se presentaron en meiosis II.
Investigaciones posteriores sugieren que el origen de la trisomia 18 es la no disyunción de los cromosomas durante la meiosis o mitosis postcigotica. Durante la investigación se observo que alrededor del 50% de los errores en la separación de los cromosomas en la ovogénesis, se presentaron en meiosis II.
Las
causas de la no disyunción de los cromosomas, se sigue investigando.
Hasta el momento se esta relacionando con polimorfismos maternos en
enzimas foliadas del metabolismo.
Síntomas
Se caracteriza por bajo peso al nacer, talla corta, retraso mental e hipertonía.

a) Anomalías cráneo faciales: cabeza pequeña, con occipucio prominente, implantación baja de las orejas, hipoplasia mandibular, cuello corto, boca pequeña, paladar ojival, labio y paladar hendido.

a) Anomalías cráneo faciales: cabeza pequeña, con occipucio prominente, implantación baja de las orejas, hipoplasia mandibular, cuello corto, boca pequeña, paladar ojival, labio y paladar hendido.
b) Anomalías oculares: ojos anormalmente pequeños, coloboma (fisura congénita en alguna parte del ojo) de iris, opacidad corneal, y cataratas.
c) Anomalías cardíacas: con comunicación anormal entre los ventrículos del corazón, afectación valvular múltiple, coartación de aorta, transposición de grandes vasos, estenosis pulmonar, tabique interventricular defectuoso, desarrollo exagerado del ventrículo derecho.

d) Anomalías esqueléticas: dedos de las manos montados, hipoplasia o aplasia radial, sindactilia, escoliosis.
e) Malformaciones urogenitales:
riñón en herradura, ectopia (estado de un órgano o tejido, situado
fuera de su lugar habitual) renal, hidronefrosis (acumulo anormal de
orina en los riñones), riñón poliquístico, criptorquidia (ausencia de
uno o ambos testículos).
Estos niños padecen infecciones frecuentes tipo neumonía, otitis media, e infecciones urinarias, y dificultades para la alimentación que incluso pueden precisar alimentación por sonda.
Estos niños padecen infecciones frecuentes tipo neumonía, otitis media, e infecciones urinarias, y dificultades para la alimentación que incluso pueden precisar alimentación por sonda.
Diagnóstico
Dado
la alta tasa de mortalidad postnatal de esta enfermedad genética, no
existe a la fecha un tratamiento. El trabajo clínico se restringe a el
diagnostico prematuro para poder supervisar el embarazo de forma
adecuada y a la preparación psicológica de los padres para una eventual
muerte perinatal inminente o el retraso mental y las incapacidades
físicas en los escasos sobrevivientes.

El diagnostico se realiza entre la semana 12 y 20 del embarazo mediante técnicas ultrasonográficas, ante la presencia de cualquier malformación que presuma una aberración cromosómica. Se confirma el diagnostico mediante amniocentesis, cordocentesis o biopsias de tejido placentario.
Síndrome de Down
Definición
Es un trastorno genético causado por la presencia de una copia extra del cromosoma 21, en vez de los dos habituales (trisomía del par 21), caracterizado por la presencia de retraso mental y rasgos físicos peculiares. Debe su nombre a John Langdon Haydon Down que fue el primero en describirlo en 1866. Este síndrome es la causa más frecuente de discapacidad psíquica congénita y afecta aproximadamente uno de cada 800 bebés.
Causas
El síndrome de Down es causado por la presencia de material genético extra del cromosoma 21. Este material extra se puede deber por tres razones:
a) Trisomía libre:
dada el 95% de los casos, donde ocurre un error durante la primera
división meiótica por la disyunción incompleta del material genético de
uno de los progenitores.
b) Translocación: ocurre
cuando una parte del cromosoma 21 se desprende durante la división
celular y se adhiere a otro cromosoma (normalmente a alguno del par 14).
Este tipo de accidente en la división celular es responsable de
aproximadamente del 3%-4% de los casos de síndrome de Down.
c) Mosaicismo: en
este caso, el accidente en la división celular tiene lugar después de
la fertilización. Las personas afectadas tienen algunas células con un
cromosoma 21 adicional y otras con la cantidad normal. Afectan 1%-2% de
los casos.

Síntomas
La persona afectada puede presentar:

* Retraso mental en diversos grados
* Baja estatura
* Dermatoglifos atípicos
* Diástasis de músulos abdominales
* Disminución del tono muscular
* Braquiocefalia (región occpital plana)
* Paladar ojival
* Manos, orejas y cuello corto
* Puente nasal deprimido
* Cardiopatía congénita
* Mayor riesgo de desarrollar patologías como hipotiroidismo, diabetes, miopía, leucemia, problemas de audición, etc.

* Retraso mental en diversos grados
* Baja estatura
* Dermatoglifos atípicos
* Diástasis de músulos abdominales
* Disminución del tono muscular
* Braquiocefalia (región occpital plana)
* Paladar ojival
* Manos, orejas y cuello corto
* Puente nasal deprimido
* Cardiopatía congénita
* Mayor riesgo de desarrollar patologías como hipotiroidismo, diabetes, miopía, leucemia, problemas de audición, etc.
Epidemiología
La incidencia global del síndrome de Down se
aproxima a uno de cada 700 nacimientos, pero el riesgo varía con la edad
de la madre. La incidencia en madres de 25 años es de 1 por cada 2000
nacidos vivos, mientras que en madres de 35 años es de 1 por cada 200
nacimientos y de 1 por cada 40 en las mujeres mayores de 40 años. Por
este motivo se recomiendan técnicas de diagnóstico prenatal a todas las
mujeres a partir de los 35 años.
La
probabilidad de tener un hijo con síndrome de Down es mayor a la media
para aquellos padres que ya han tenido otro previamente. Típicamente la
probabilidad de tener otro hijo con el síndrome en cada embarazo
subsiguiente es de una por cada cien recién nacidos vivos. Los
antecedentes familiares igualmente incrementan ese riesgo.
Los varones con síndrome de Down se consideran estériles, pero las mujeres conservan con frecuencia su capacidad reproductiva. En su caso también se incrementa la probabilidad de engendrar hijos con el síndrome hasta un 50%, aunque pueden tener hijos sin trisomía.
Los varones con síndrome de Down se consideran estériles, pero las mujeres conservan con frecuencia su capacidad reproductiva. En su caso también se incrementa la probabilidad de engendrar hijos con el síndrome hasta un 50%, aunque pueden tener hijos sin trisomía.
Diagnóstico
Existen varias formas de determinar el síndrome de Down, entre ellas están la “triple prueba”, la más común y más utilizada, consiste en pruebas de AFP (Alfa-fetoproteína), estriol y hCG (Gonadotropina coriónica humana) para la detección de niveles anorma
 les que indiquen el padecimiento del síndrome.
les que indiquen el padecimiento del síndrome.Otras formas de diagnosticar el síndrome de Down están ecografías (longitud del fémur, grosor del pliegue nucal, etc.), conteo cromosómico a través de alguna célula fetal conseguida con técnicas como amniocentesis y biopsia de vellosidades coriónicas, etc.
Tratamiento
La mejoría en los tratamientos de las enfermedades
asociadas al síndrome de Down ha aumentado la esperanza de vida de estas
personas, entre los 50 años y los 60 años. A lo largo de los últimos
años se han postulado diferentes tratamientos prácticos (hormona
tiroidea, hormona del crecimiento, complejos vitamínicos y minerales,
etc.) sin que ninguno haya demostrado ningún efecto positivo
significativo en el desarrollo motor, social, intelectual de las
personas con el síndrome de Down.
Los únicos tratamientos que han demostrado una influencia significativa en el desarrollo de los niños con síndrome de Down son los programas de Atención Temprana, orientados a la estimulación precoz del sistema nervioso central durante los seis primeros años de vida. Los individuos con grandes dificultades para el aprendizaje a menudo han sido internados en instituciones, pero se ha comprobado que deben vivir en su domicilio, donde desarrollan de forma más completa todo su potencial. Cuando éste es demasiado protector, los chicos y chicas tienden a dejarse llevar, descubriendo escasamente sus potencialidades. Los contextos estimulantes ayudan a que se generen conductas de superación que impulsan el desarrollo de la inteligencia.

Síndrome de Patau
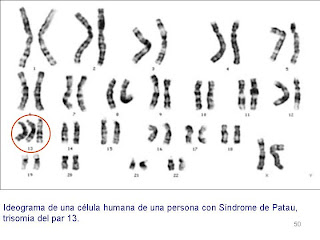
Definición
El síndrome
de Patau o trisomía del cromosoma 13 es una enfermedad cromosómica rara
caracterizada por la presencia de un cromosoma 13 adicional. Fue
descrita por primera vez por Patau en 1960.
Causas
La trisomía 13 puede darse por:
a) Presencia de un cromosoma 13 extra (tercer cromosoma) en todas las células.
b) Mosaicismo, presencia de un cromosoma 13 extra en algunas de las células.
c) Trisomía parcial, presencia de una parte de un cromosoma 13 extra en las células.
a) Presencia de un cromosoma 13 extra (tercer cromosoma) en todas las células.
b) Mosaicismo, presencia de un cromosoma 13 extra en algunas de las células.
c) Trisomía parcial, presencia de una parte de un cromosoma 13 extra en las células.
Es la trisomía más rara con una prevalencia de uno en 12.000 nacidos vivos y predomina ligeramente en el sexo femenino.
Al
igual que en otras trisomías de mayor difusión, como en el síndrome de
Down, la edad materna y paterna es un factor de riesgo y en estos casos
suelen ser superiores a los 30 años.
 Diagnóstico y pronóstico
Diagnóstico y pronóstico
Para diagnosticar este síndrome se puede hacer
mediante la ecografía, que consta un retraso en el crecimiento
intrauterino y pueden ser detectadas algunas de las anomalías que
caracterizan este síndrome, especialmente la holoprosencefalia (ausencia
de desarrollo de lo que será el lóbulo frontal del cerebro) y las
distintas malformaciones renales, cardiacas y faciales. En los casos en
que se sospeche este síndrome será obligada la amniocentesis para
dilucidar el cariotipo fetal.
El
pronóstico vital es muy grave; la inmensa mayoría de estos enfermos
fallecen de muy pocas semanas de edad, debido a los problemas
cardiorrespiratorios.
Síntomas

* Labio leporino
* Ojos muy juntos: los ojos realmente pueden fusionarse en uno
* Polidactilia
* Retardo psicomotor y mental
* Fisura congénita en el iris del ojo (coloboma)
* Cardiopatías congénitas
* Anomalías diafragmáticas, urogenitales y sensoriales
* Disminución del tono muscular
* Microcefalia y micrognacia
* Testículo no descendido (criptorquidia)
* Riñones poliquísticosTratamiento
Los
recien nacidos con trisomía 13 suelen necesitar asistencia médica desde
el mismo momento del nacimiento debido a que 2/3 de los casos obtienen
puntuaciones inferiores a 7 en el test de Apgar al primer minuto, cifra
que desciende a 1/3 a los 5 minutos de vida. Dado que las anomalías
cardiacas representan la causa principal de morbimortalidad, se plantea
el problema ético de si su recuperación quirúrgica está indicado dado el
pésimo pronóstico del cuadro tanto desde el punto de vista físico como
intelectual. Alrededor de 2/3 de los pacienets son dados de alta y
precisan de atención especializada en el domicilio, requiriendo la
intervención de un equipo multidisciplinar. Los padres han de ser
previamente entrenados para la realización de determinadas tareas y
maniobras que pueden ser de importancia vital para la supervivencia del
recien nacido.
Alteraciones cromosómicas estructurales
Estas anomalías afectan a la estructura del cromosoma en cuanto a la
ordenación lineal de los genes. Uno o más cromosomas cambian su
estructura propia por la adición o pérdida de material genético, por
alteración de su forma o del patrón de bandas. Estos cambios se llaman
reorganizaciones y siempre se relacionan con rotura cromosómica. Aquí se
incluyen las siguientes anomalías.
Deleciones
Consiste en la pérdida de un fragmento del cromosoma, lo que origina un
desequilibrio (el portador de una deleción es monosómico respecto al
alelo afectado por la deleción). Se puede producir en el extremo de un
cromosoma (deleción terminal) o a lo largo de sus brazos corto o largo
(deleción intersticial). Una de las causas del síndrome de Prader-Willi es una deleción parcial del brazo largo del cromosoma 15.
Duplicaciones
se trata de la duplicación de una región cromosómica concreta, por lo que su portador tendrá material genético extra. El síndrome de X frágil es debido a una duplicación parcial del extremo del brazo largo del cromosoma X.
Inversiones
un segmento del cromosoma cambia su orientación dentro del cromosoma, cambia de sentido. Existen dos tipos de inversiones: Pericéntrica, cuando el centrómero forma parte del segmento invertido, o Paracéntrica en el caso contrario. Tienen una frecuencia muy baja y no suelen causar grandes trastornos.
Cromosoma en anillo
Se presenta cuando ambos brazos de un cromosoma se fusionan formando un
anillo. Aunque son poco frecuentes, también están implicados en
enfermedades. Por ejemplo, una de las causas del síndrome de Turner es la formación de una anillo en el cromosoma X.
Translocaciones
Tienen lugar cuando una porción de un cromosoma se transfiere a otro
cromosoma. Cuando un segmento se intercambia entre dos cromosomas no
homólogos tiene lugar una translocación recíproca. Se produce una
reordenación del material genético, pero no hay pérdida o ganancia de
información genética como en el caso de las deleciones y las
duplicaciones. La translocación robertsoniana es una caso especial de translocación ("casi equilibrada)
en el que dos cromosomas no homólogos pierden sus brazos cortos
mientras que los largos se unen por el centrómero de uno de los
cromosomas, formándose un cromosoma único. Este tipo de translocación
afecta a los cromosomas acrocéntricos con el brazo p muy pequeño (en
humanos, los cromosomas 13, 14, 15, 21 y 22). El 4% de los casos de síndrome de Down se deben a una translocación 21/21 o 14/21.
Inserción
Consiste en la inserción de un segmento de ADN en un lugar diferente,
lo que puede dar como resultado la alteración de la estructura y función
normales de un gen.
Isocromosoma
es un cromosoma que ha perdido un brazo y el otro se ha duplicado, de
modo que existe una monosomía parcial debido al brazo perdido, y a una
trisomía parcial, debido al brazo duplicado.
Bibliografía
Robert L., Nussbaum; Roderick R. McInnes; Huntington F. Willard (2008). «Capítulo 5: Principios de citogenética clínica» (en castellano). Thompson & Thompson. Genética en Medicina (7ª edición). Barcelona: Elsevier Masson. pp. 68-75. ISBN 978-84-458-1870-1.Tipos de anomalias
Anomalías numéricas
Estas anomalías se denominan también mutaciones genómicas, ya que varía el número de cromosomas del genoma. Pueden ser aneuploidías o poliploidías. El caso más común es la aneuploidía, que se produce cuando un individuo presenta accidentalmente algún cromosoma de más (trisomía, 2n+1) o de menos (monosomía, 2n-1) en relación con su condición normal (diploide)
Las poliploidías se producen cuando se tiene tres o más juegos completos de cromosomas (Triploidía,3n;
Tetraploidía, 4n). En humanos, las triploidías suelen acabar en aborto y
si se llega al nacimiento, termina sufriendo una muerte prematura. La
tetraploidía es letal.
Aneuploidías autosómicas
Son alteraciones en el número de copias de alguno de los cromosomas
no sexuales. En humanos, no todas las aneuploidías numéricas son
viables, y las que sí lo son producen alteraciones en el fenotipo. Entre
las más frecuentes destacan:
- Trisomía del cromosoma 21 más conocida como Síndrome de Down (es la causa del 95% de los casos).
- Trisomía del cromosoma 18 más conocida como Síndrome de Edwards.
- Trisomía del cromosoma 13 más conocida como Síndrome de Patau.
- Trisomía del cromosoma 22 (letal, se han descrito casos de mosaicismo).
- Monosomía del cromosoma 21 (letal, se han descrito casos de mosaicismo).
Variaciones numéricas en cromosomas claves en el desarrollo temprano
del embrión no son viables ni siquiera a nivel embrionario, por lo que
no se detectan como causas frecuentes de abortos espontáneos causados
por aneuploidías cromosómicas (es el caso del cromosoma 1, por ejemplo).
Aneuploidías sexuales
Son alteraciones en el número de copias de alguno de los dos cromosomas sexuales humanos. Las aneuploidías en este caso suelen ser viables. Entre las más frecuentes destacan:
- Síndrome de Klinefelter (trisomía de los cromosomas sexuales: 47, XXY).
- Síndrome de Turner (monosomía de los cromosomas sexuales: 45, X). Es la única monosomía viable.
- Síndrome del doble Y (llamado a veces síndrome del supermacho: 47, XYY).
- Síndrome del triple X (llamado a veces síndrome de la superhembra: 47, XXX).
Bibliografía
Robert L., Nussbaum; Roderick R. McInnes; Huntington F. Willard (2008). «Capítulo 5: Principios de citogenética clínica» (en castellano). Thompson & Thompson. Genética en Medicina (7ª edición). Barcelona: Elsevier Masson. pp. 68-75. ISBNEscrito malformaciones geneticas http://www.monografias.com/trabajos-pdf/malformaciones-congenitas/malformaciones-congenitas.shtml
Introduccion a las enfermedades geneticas

Son enfermedades relacionadas a las
alteraciones o anomalías de los cromosomas, en el cual, el afectado
sufre un desorden en el material genético. Se hablara de que se tratan,
síntomas, tratamiento, etc.
¿que es una mutacion cromosomica?
son alteraciones en el número o en la estructura de los cromosomas.
¿a que se deben?
a errores durante la gametogénesis (formación de los gametos por meiosis) o de las primeras divisiones del cigoto. En el primer caso la anomalía
estará presente en todas las líneas celulares del individuo, mientras
que cuando la anomalía se produce en el cigoto puede dar lugar a mosaicismo, coexistiendo por tanto poblaciones de células normales con otras que presentan mutaciones cromosómicas.
¿cuando pueden ser observadas estas mutaciones?
pueden ser observadas durante la metafase del ciclo celular y que tienen su origen en roturas (procesos clastogénicos) de las cadenas de ADN no reparadas o mal reparadas, entre otros factores.
¿que es una aneuploidia?
hace referencia al cambio en el número cromosómico, que pueden dar lugar a enfermedades genéticas.Un aneuploide es un individuo cuyo número de cromosomas difiere del tipo salvaje o euploide en parte de su dotación cromosómica, debido a un cromosoma extra o ausente, que siempre se asocia con una deficiencia en el desarrollo físico, mental o ambos. Generalmente, la dotación cromosómica aneuploide sólo difiere de la salvaje en uno o pocos cromosomas.¿donde se pueden observar comunmente las aneuploidias?
se puede observar frecuentemente en células cancerosas. En los animales sólo son viables las monosomías y las trisomías, ya que las nulisomías son letales en individuos diploides.
el fenómeno por el cual se originan células, tejidos u organismos con tres o más juegos completos de cromosomas de la misma o distintas especies o con dos o más genomas de especies distintas. Tales células, tejidos u organismos se denominan poliploides.
¿Donde se pueden observar comúnmente las poliploidias?
 es más frecuente en plantas y algas que en animales y hongos. En plantas, la poliploidía se encuentra muy extendida dentro de las angiospermas (aproximadamente un 30% de las especies son alopoliploides) y parece estar relacionada con la latitud geográfica.
es más frecuente en plantas y algas que en animales y hongos. En plantas, la poliploidía se encuentra muy extendida dentro de las angiospermas (aproximadamente un 30% de las especies son alopoliploides) y parece estar relacionada con la latitud geográfica.
¿Que es una poliploidia?
el fenómeno por el cual se originan células, tejidos u organismos con tres o más juegos completos de cromosomas de la misma o distintas especies o con dos o más genomas de especies distintas. Tales células, tejidos u organismos se denominan poliploides.
¿Donde se pueden observar comúnmente las poliploidias?
Sabias que...
Actualmente se dispone de un amplio conocimiento del cariotipo
humano y de las anomalías cromosómicas. Puesto que estas alteraciones
son anomalías genéticas, pueden transmitirse a la descendencia en el
caso de que afecten a las células germinales.
Sabias que...
Se estima que cerca de un 60% de los abortos ocurridos en el primer
trimestre de gestación se deben a anomalías cromosómicas y un 0,5% de
los recién nacidos presentan aneuploidías.Por este motivo, el estudio de estas mutaciones mediante un cariotipo o un FISH es de gran utilidad para detectar anticipadamente cualquier anomalía.
Bibliografia
Bibliografía
Robert L., Nussbaum; Roderick R. McInnes; Huntington F. Willard (2008). «Capítulo 5: Principios de citogenética clínica» (en castellano). Thompson & Thompson. Genética en Medicina (7ª edición). Barcelona: Elsevier Masson. pp. 68-75. ISBN
Suscribirse a:
Entradas (Atom)